This document is © Elsevier Science, 1994
Planarity of conjugated cyclic systems
and prediction of rotational constants:
ab initio and semi-empirical calculations
on some cross-conjugated compounds
*Abteilung Chemische Physik,
University of Ulm, D-89069 Ulm (Germany)
**Abteilung Elektrochemie,
University of Ulm,
D-89069 Ulm (Germany)
Abstract
Ab initio and semi-empirical calculations have been carried out on six
cyclic cross-conjugated compounds: fulvene, cyclopentadienone,
2,5-cyclohexadienone, 4,4-dimethyl-2,5-cyclohexadienone,
3-methylene-1,4-cyclohexadiene and 3,3-dimethyl-6-methylene-1,4-
cyclohexadiene. The results were compared with experimental data as far as
available.
Introduction
The most important molecular quantities for rotational spectroscopy are
the rotational constants of a molecule. They are defined as
Bg = h/(8p²·Ig), g = a, b, c
where Ig is the moment of inertia with respect to the principal inertial
axis g. The moment of inertia is given by
Ig = S(mi·rgi²)
where mi signifies the mass of atom i and rgi the distance of the ith atom
from the g axis.
Therefore, to predict the rotational spectrum of a certain molecule - which
is an almost indispensable prerequisite for assigning the spectrum -
without having relevant data available from other methods one needs to
estimate the molecular geometry of the molecule.
In the course of our investigations in the field of cyclic cross-conjugated
compounds predictions for rotational constants had to be made frequently.
Since no useful experimental data were available for most of the molecules,
we had to investigate the molecular geometries of these compounds
theoretically.
There are two basic approaches for such an investigation. First, one can
adapt parameters from similar molecules that have been determined
experimentally assuming them to be the same in the molecule under study.
As there is a considerable bandwidth for most parameters, the rotational
constants obtained by this method are unlikely to be accurate enough in
most cases.
With the advent of fast computers a second possibility has become feasible:
the calculation of geometries by empirical forcefield or quantum mechanical
methods. The accuracy of these geometries - and the resulting rotational
constants - is the objective of this paper.
Planarity
Besides being able to predict microwave spectra we had a particular
interest in the question whether the molecules under study are planar. In
the case of five-membered cross-conjugated rings this question can be - and
indeed has been - answered by microwave spectroscopy rather easily by just
determining the three principal moments of inertia and then calculating the
defect of inertia D, which is defined as
D = Ic - Ia - Ib
and should be close to zero for planar molecules. The results were
planarity for both cyclopentadienone (Ia)
[1,2] and the methylene analog
fulvene (Ib) [3,4].
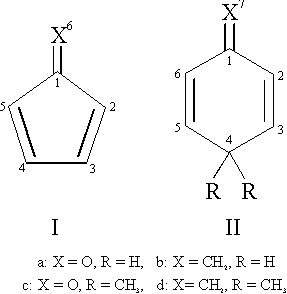
The situation with six-membered rings is more complex. The carbon atom in
4-position shows sp³-hybridization and so there are always at least
two atoms out of the ring plane. If the out-of-plane atoms are just two
hydrogens the defect of inertia can be evaluated in good approximation.
The corresponding molecules (IIa and IIb), however,
are not stable. We were unable to detect 2,5-cyclohexadienone (IIa) by microwave spectroscopy and the works on this molecule
that have been published so far [5,6] do not include
any structural information. The methylene analog 1-methylene-2,5-
cyclohexadiene1 (IIb) has been known for almost
three decades [7-9], but no structural information
was available until recently, when the molecule was shown to have a planar
ring by microwave spectroscopy [10].
A main reason for the instability of the two compounds
(IIa) and (IIb) is tautomerism. The carbonyl compound rearranges to
form phenol, while the methylene compound mainly forms toluene. One
possibility to prevent this rearrangement is to substitute both hydrogens
in 4-position by methyl groups. In the case of the ketone this leads to
4,4-dimethyl-2,5-cyclohexadienone (IIc), which was
also investigated by microwave spectroscopy [11].
The two methyl groups, however, make a decision about planarity impossible
on the basis of the rotational constants of one isotopomer alone. The
structure of the corresponding methylene compound (
IId) has been investigated by electron diffraction
[12] and the data were interpreted as indicative of a non-planar ring
with dihedral angles within the ring of about 8 degrees.
Theoretical calculations
We thought it would be worthwhile comparing the structural data we obtained
from the experiments with those predicted by theoretical calculations. Such
a comparison would provide information about the reliability of theoretical
results that can not yet be checked by experimental values. So we calculated
the structures of several cross-conjugated molecules with ab-initio methods
at two different levels of theory (RHF-6/31G* and STO-3G) using GAUSSIAN86
[13] and also semi-empirically with three different
Hamiltonians (MINDO/3 [14a]], MNDO [14b] and AM1 [14c]) using MOPAC [14d]. The calculations were carried out using the DEC
VAX 9410/6440 cluster of the computational center of the University
of Ulm. All the molecules considered in this study possess CS symmetry
if they are non-planar, whereas the symmetry is C2v if they are planar. So
we started with only CS symmetry imposed on the calculations.
2,5-Cyclohexadienone (cf. Table 1)
The initial parameters for cyclohexadienone (IIa)
were taken from an optimized structure calculated by the molecular modeling
package SYBYL [15a] using a TRIPOS-forcefield
[15b]. The structure calculated by SYBYL
was planar no matter if the starting-conformation was planar, boat, chair or
half-chair. So we twisted the molecule arbitrarily to a conformation where
the C6-C1-C2-plane and the C3-C4-C5-plane were at plane angles of 2 degrees
and 10 degrees respectively to the C2-C3-C5-C6-plane. This conformation was
then used as the initial conformation for the optimizations with only CS
symmetry fixed.
All optimized geometries were planar within the convergence criteria and so
we decided to restrain the molecule to the indicated C2v symmetry for a
conformation that would be exactly planar without having to increase
computing time overly.
4,4-Dimethyl-2,5-cyclohexadienone (cf. Table2)
For the dimethylated ketone (IIc) we chose the C2v
optimized geometry of (IIa) with reasonable
parameters for the methyl groups, but with only CS symmetry required. The
resulting structures, again, were planar without doubt and so the same
procedure as in the case of (IIa) was applied in
order to obtain values for an exactly planar geometry.
Methylene compounds with six membered rings (cf. Tables 3 and 4)
In the case of the methylene-compounds (IIb and
IId) the initial parameters used were those given in an electron
diffraction study by Trætteberg et al. [12] for
(IIb) (i.e. dihedral angles within the ring of
about 8 degrees). However all optimized structures were found to be planar
with fixed CS symmetry and thus were refined with fixed C2v symmetry, too.
Cyclopentadienone and fulvene (cf. Tables 5 and 6)
The initial parameters for both molecules were taken from the geometry of
fulvene as determined by Baron et al. [4] and Suenram
et al. [16]. For cyclopentadienone a value of 1.22
Å for the C=O bond length was used. As with the other molecules only
CS symmetry was imposed and only after the calculations had converged to a
planar structure these values were refined with C2v symmetry.
Discussion
The same result, that is planar rings for all compounds, was obtained from
ab-initio, semi-empirical calculations and by calculations using the TRIPOS
forcefield. This is in agreement with the experimental results as far as
they are available, except for 4,4-dimethyl-1-methylene-2,5-cyclohexadiene.
However, considering the fact that 1-methylene-2,5-cyclohexadiene was
unambiguously shown to have a planar ring [10],
there is no reason to expect a non-planar ring structure for the
dimethylated compound, and the interpretation of the experimental data
given in [12] probably is in error.
A survey of the obtained deviations from experimental results is given in
Table 7. Reliable values for bond lengths and
angles are available only for fulvene. For this molecule the MNDO method
gives the best agreement with the experimental results (average deviation
for all distances given in Table 6: Drrms =
0.8%, average deviation for all angles given in Table
6: Darms = 1.43%), but RHF/6-31G* (Drrms = 1.02%,
Darms = 1.49%) and AM1 (Drrms = 1.08%, Da
rms = 1.57%) also agree quite well. Regarding the rotational constants of
fulvene, however, the situation is different: best agreement is achieved
with STO-3G (average deviation of the three rotational constants: D
Brms = 0.62%). AM1 (DBrms = 0.88%) and RHF/6-31G* (DBrms =
1.22%) still agree well, but MNDO constants show a larger deviation
(DBrms = 1.92%).
Similar results are obtained for the other molecules where experimentally
determined rotational constants are available. In three of four cases the
best agreement is achieved with STO-3G, RHF/6-31G* or AM1. MINDO/3 agrees
worst in these cases whereas it gives the best agreement among the
semi-empirical methods for cyclopentadienone.
In summary it can be said that for the cross-conjugated compounds
considered in this study, the overall agreement of the predicted rotational
constants with the experimentally determined ones is quite good for STO-3G,
RHF/6-31G* and AM1. Whereas the rotational constants determined by MNDO and
MINDO/3 show deviations that are almost twice as high. The exact "ranking"
differs depending on the molecule that is studied. Taking into account the
amount of time used for the computations - several hours to days for ab
initio calculations compared to some seconds to a few minutes for
semi-empirical calculations - it seems that for predicting rotational
constants of cross-conjugated cyclic compounds the AM1 method is the most
efficient one.
The predicted constants for 2,5-cyclohexadienone, which could not be
measured up to now, should be correct within ca. 1.5%. In particular the
ring in the molecule should be expected to be planar.
Note added in proof
Additional information on this work is contained in the dissertation thesis of W.H.
(in German).
References
1 P.A. Baron and R.D. Brown, Chem. Phys. 1 (1973) 444.
2 a) K. Bestle and H.-K. Bodenseh, 12th Coll. High Res. Mol. Spec. Dijon
(1991) F12.
b) R. Ruoff and H.-K. Bodenseh, 13th Coll. High Res. Mol. Spec. Riccione
(1993) F11.
3 R.D. Brown, F.R. Burden and J.E. Kent, J. Chem. Phys. 49 (1968) 5542.
4 P.A. Baron, R.D. Brown, F.R. Burden, P.J. Domaille and J.E. Kent,
J. Mol. Spectrosc., 43 (1972) 401.
5 M.-C. Lasne, J.-L. Ripoll and J.-M. Denis, Tetr.Lett. 21 (1980) 463.
6 C.S. Shiner, P.E. Vorndam and S.R. Kass, J.Am.Chem.Soc. 108 (1986)
5699.
7 H. Plieninger and W. Maier-Borst, Chem. Ber., 98 (1965) 2504.
8 J.J. Gajewski and A.M. Gortva, J. Am. Chem. Soc., 104 (1982) 334.
9 J.E. Bartmess, J. Am. Chem. Soc., 104 (1982) 335.
10 W. Hutter, H.-K. Bodenseh and A. Koch, J. Mol. Struct., 319 (1994)
73.
11 W. Hutter and H.-K. Bodenseh, J. Mol. Struct., 291 (1993) 151.
12 M. Trætteberg, P. Bakken, A. Almenningen, W. Lüttke and J. Janssen,
J. Mol. Struct. 81 (1982) 87.
13 M.J. Frisch, J.S. Binkley, H.B. Schlegel, K.Raghavachari, C.F.
Melius, R.L. Martin, J.J.P. Stewart, F.W. Bobrowicz, C.M. Rolfing,
L.R. Kahn, D.J. Defrees, R. Seeger, R.A. Whiteside, D.J. Fox, E.M.
Fleuder and J.A. Pople, GAUSSIAN86, Carnegie-Mellon Quantum Chemistry
Publishing Unit, Pittsburgh PA, 1984.
14 a) M.J.S. Dewar, R.C. Bingham and D.H. Lo, J. Am. Chem. Soc. 97 (1975)
1285.
b) M.J.S. Dewar and W. Thiel, J. Am. Chem. Soc. 99 (1977) 4899.
c) M.J.S. Dewar, E.G. Zoebisch, E.F. Healy and J.J.P. Stewart, J. Am.
Chem. Soc. 107 (1985) 3902.
d) M.J.S. Dewar, J. Mol. Struct. 100 (1983) 41.
15 a) SYBYL 5.2., Tripos Inc., 1699 S. Hanley Road, Suite 303,
St. Louis, MO, 63144, (1988).
b) M. Clark, R.D. Cramer III and N. Van Opdenbosch, J. Comp. Chem. 10
(1989) 982.
16 R.D. Suenram and M.D. Harmony, J. Chem. Phys. (1973) 5842.
TABLE 1
Molecular structure* of cyclohexadienone (IIa) as
obtained from ab-initio and semi-empirical calculations.
_______________________________________________________________
GAUSSIAN86 MOPAC
6-31G* STO-3G MNDO MINDO/3 AM1
_______________________________________________________________
C1=O 1.199 1.227 1.228 1.207 1.239
C1-C2 1.482 1.511 1.498 1.508 1.474
C2=C3 1.324 1.317 1.349 1.349 1.339
C3-C4 1.499 1.520 1.506 1.498 1.483
C2-H 1.075 1.083 1.092 1.106 1.102
C3-H 1.077 1.085 1.093 1.108 1.102
C4-H 1.090 1.093 1.116 1.119 1.127
C2-C1=O 121.8 122.5 122.3 122.8 122.2
C2-C1-C6 116.4 114.9 115.5 114.4 115.6
C1-C2=C3 121.7 122.3 121.9 122.5 122.0
C2=C3-C4 123.3 123.7 123.6 123.5 123.3
C3-C4-C5 113.8 113.1 113.5 113.7 113.8
C1-C2-H 116.1 116.2 117.0 118.0 115.8
C3=C2-H 122.3 121.5 121.0 119.5 122.2
C2=C3-H 120.1 120.7 121.0 121.2 121.3
C4-C3-H 116.6 115.6 115.4 115.4 115.5
C3-C4-H 109.4 109.4 109.2 110.0 109.0
H-C4-H 105.1 106.0 106.4 102.8 106.8
A 5235.36 5128.09 5174.67 5198.95 5306.56
B 2721.84 2648.84 2615.96 2620.38 2650.41
C 1810.15 1765.36 1757.00 1760.95 1788.23
µ 4.7 2.8 3.5 4.2 4.2
________________________________________________________________
*Distances are in Å, angles in degrees, rotational constants in MHz,
dipole moments in Debye.
TABLE 2
Molecular structure* of 4,4-dimethyl-2,5-cyclohexadien-1-one (IIc) as obtained from ab-initio and semi-empirical
calculations compared with experimental data.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.** [11]
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=O 1.198 1.227 1.228 1.207 1.238
C1-C2 1.481 1.509 1.495 1.497 1.473
C2=C3 1.323 1.313 1.348 1.348 1.338
C3-C4 1.508 1.530 1.526 1.538 1.498
C2-H 1.075 1.083 1.092 1.106 1.102
C3-H 1.078 1.085 1.094 1.110 1.102
C4-C7 1.543 1.557 1.557 1.536 1.528
C7-H' 1.084 1.086 1.109 1.111 1.116
C7-H'' 1.085 1.086 1.109 1.111 1.116
C2-C1=O 122.0 122.7 122.5 123.3 122.3
C2-C1-C6 116.0 114.6 114.9 113.4 115.5
C1-C2=C3 121.6 122.4 122.3 123.0 122.2
C2=C3-C4 124.7 124.7 124.8 126.4 124.0
C3-C4-C5 111.5 111.2 110.8 107.9 112.3
C3=C2-H 122.2 121.5 120.7 118.8 122.0
C2=C3-H 119.7 120.3 119.7 119.3 121.3
C3-C4-C7 109.0 109.1 108.8 109.6 108.8
C4-C7-H' 110.8 110.6 111.2 113.5 110.4
C4-C7-H" 110.7 110.4 111.5 113.4 110.0
H'-C7-H" 108.1 108.4 107.6 105.2 108.8
H"-C7-H" 108.2 108.6 107.4 105.3 109.0
C7-C4-C8 109.3 109.1 110.8 110.4 109.2
A 3338.80 3276.33 3260.03 3306.67 3370.32 3332.16
B 1205.23 1178.95 1165.69 1151.86 1197.24 1193.07
C 1092.49 1067.37 1061.41 1050.04 1084.41 1082.21
µ 4.8 2.9 3.6 4.3 4.2 4.5
____________________________________________________________________________
*Distances are in Å, angles in degrees, rotational constants in MHz,
dipole moments in Debye. **The experimental error is less than the unit of
the last digit given.
TABLE 3
Molecular structure* of 1-methylene-2,5-cyclohexadiene
(IIb) as obtained from ab initio and semi-empirical calculations
compared with experimental data.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.** [10]
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=C7 1.328 1.320 1.353 1.344 1.343
C1-C2 1.473 1.492 1.479 1.496 1.459
C2=C3 1.323 1.314 1.351 1.349 1.341
C3-C4 1.503 1.521 1.505 1.497 1.484
C2-H 1.076 1.083 1.093 1.108 1.102
C3-H 1.077 1.084 1.092 1.107 1.101
C4-H 1.090 1.093 1.116 1.120 1.127
C7-H 1.075 1.081 1.089 1.100 1.097
C2-C1=C7 122.2 122.4 122.4 123.6 122.0
C2-C1-C6 115.6 115.1 115.2 112.8 115.9
C1-C2=C3 122.4 122.6 122.7 123.8 122.2
C2=C3-C4 123.8 123.5 123.3 123.2 123.2
C3-C4-C5 112.8 112.5 112.9 113.2 113.5
C1-C2-H 117.3 116.6 117.2 116.4 116.5
C3=C2-H 120.3 120.7 120.2 119.8 121.3
C2=C3-H 119.8 120.6 121.0 120.7 121.3
C4-C3-H 116.8 115.8 115.6 116.0 115.6
C3-C4-H 109.7 109.6 109.4 110.2 109.1
H-C4-H 105.1 105.7 106.2 102.4 106.8
C1-C7-H 121.7 121.9 123.4 125.0 122.2
H-C7-H 116.7 116.2 113.1 110.0 115.6
A 5194.13 5100.58 5167.70 5181.15 5254.47 5177.82
B 2645.62 2634.14 2559.81 2523.74 2621.12 2613.15
C 1771.37 1755.54 1730.67 1714.78 1768.95 1755.84
µ 0.8 0.6 0.1 0.1 0.5 0.86
____________________________________________________________________________
*Distances are in Å, angles in degrees, rotational constants in MHz,
dipole moments in Debye. **The experimental error is less than the unit of
the last digit given.
TABLE 4
Molecular structure* of 1-methylene-4,4-dimethyl-2,5-cyclohexadiene (IId) as obtained from ab initio and semi-empirical
calculations compared with experimental data.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp. [12]
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=C7 1.328 1.320 1.353 1.344 1.343 1.357**
C1-C2 1.472 1.491 1.477 1.493 1.459 1.478(6)
C2=C3 1.323 1.314 1.350 1.348 1.339 1.352(1)
C3-C4 1.512 1.530 1.525 1.539 1.498 1.493(6)
C2-H 1.077 1.083 1.093 1.108 1.102 1.075(5)
C3-H 1.078 1.084 1.093 1.109 1.101 1.075(5)
C4-C8 1.542 1.557 1.556 1.536 1.528 1.565(3)
C8-H' 1.084 1.086 1.109 1.112 1.116 1.175(4)
C8-H" 1.086 1.086 1.109 1.112 1.116 1.175(4)
C7-H 1.075 1.081 1.089 1.100 1.097 1.075(5)
C2-C1=C7 122.4 122.6 122.7 124.1 122.1
C2-C1-C6 115.3 114.8 114.6 111.8 115.7 115.7(3)
C1-C2=C3 122.4 122.7 123.1 124.3 122.4 120.8(3)
C2=C3-C4 124.6 124.5 124.5 126.1 123.7 125.6**
C3-C4-C5 110.7 110.8 110.3 107.5 112.0 110.5**
C3=C2-H 120.3 120.7 119.8 119.1 121.1 119.0**
C2=C3-H 119.5 120.3 119.8 119.0 121.4 119.5**
H'-C8-H" 108.1 108.4 107.5 105.1 108.8 110.0**
H"-C8-H" 108.1 108.5 107.3 105.0 108.9 110.0**
C8-C4-C9 109.1 109.0 110.7 110.1 109.0 112.5§
H-C7-H 116.7 116.2 113.2 110.1 115.6 121.0**§§
d$ 0.0 0.0 0.0 0.0 0.0 7.9(6)
A 3325.53 3266.14 3258.06 3300.81 3351.94 3229.59$$
B 1195.20 1185.48 1161.36 1136.31 1200.43 1183.27$$
C 1081.77 1071.21 1057.20 1035.90 1084.68 1082.05$$
µ 0.7 0.7 0.1 0.1 0.5
____________________________________________________________________________
*Distances are in Å, angles in degrees, rotational constants in MHz,
dipole moments in Debye. **No error given. §Assumed. §§Calculated from
complementary angle. $Dihedral angle C6-C1-C2=C3. $Calculated from the
structure given by ref. [12].
TABLE 5
Molecular structure* of cyclopentadienone (Ia)
as obtained from ab initio and semi-empirical calculations compared with
experimental data.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.**
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=O 1.188 1.220 1.218 1.198 1.224
C1-C2 1.506 1.525 1.518 1.521 1.509
C2=C3 1.322 1.317 1.358 1.352 1.353
C3-C4 1.503 1.506 1.489 1.494 1.490
C2-H 1.071 1.080 1.081 1.098 1.088
C3-H 1.073 1.082 1.083 1.101 1.090
C2-C1=O 127.3 128.0 127.8 127.7 127.7
C2-C1-C5 105.5 104.0 104.4 104.6 104.7
C1-C2=C3 107.5 108.1 108.2 108.0 108.3
C2=C3-C4 109.8 109.9 109.6 109.7 109.4
C1-C2-H 123.5 123.5 123.1 124.6 122.1
C3=C2-H 129.0 128.5 128.6 127.4 129.6
C2=C3-H 127.0 127.4 127.9 128.1 128.4
C4-C3-H 123.2 122.7 122.6 122.1 122.2
A 8216.82 8170.95 8239.60 8161.31 8287.97 8152.17§
B 4024.21 3902.38 3831.37 3880.45 3843.75 3939.56§
C 2701.26 2641.04 2615.28 2629.97 2625.92 2655.80§
µ 3.6 1.9 2.8 3.4 2.9 3.1§§
____________________________________________________________________________
*Distances are in Å, angles in degrees, rotational constants in MHz,
dipole moments in Debye. **The experimental error is less than the unit of
the last digit given. §From ref. [2a]. §§From ref.
[1].
TABLE 6
Molecular structure* of fulvene (Ib) as obtained
from ab initio and semi-empirical calculations compared with experimental
data.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.**
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=C6 1.325 1.319 1.345 1.338 1.332 1.3485(5)§
C1-C2 1.477 1.495 1.491 1.507 1.483 1.468(1)§§
C2=C3 1.333 1.324 1.366 1.359 1.363 1.357(1)§§
C3-C4 1.480 1.492 1.476 1.480 1.476 1.476(2)§
C2-H 1.073 1.080 1.082 1.100 1.088 1.078(1)§
C3-H 1.073 1.081 1.082 1.100 1.088 1.080(1)§
C6-H 1.076 1.083 1.089 1.101 1.098 1.077(1)§§
C2-C1=C6 127.3 127.8 127.8 128.5 127.4
C2-C1-C5 105.5 104.4 104.4 103.0 105.2 106.6(1)§
C1-C2=C3 108.2 108.6 109.0 109.7 108.6 107.7(1)§
C2=C3-C4 109.1 109.2 108.8 108.9 108.8 109.0(2)§
C1-C2_H 124.3 123.5 123.4 122.7 122.9 124.7(2)§
C3=C2-H 127.5 127.9 127.7 127.7 128.6
C2=C3-H 126.6 127.2 127.7 127.7 128.0 126.4(2)§
C4-C3-H 124.3 123.6 123.5 123.4 123.1
C1-C6-H 121.7 122.0 123.2 125.1 122.2 126.4(1)§§
H-C6-H 116.6 116.0 113.6 109.9 115.7
A 8240.05 8152.65 8223.85 8184.11 8218.53 8186.13§
B 3861.77 3835.81 3696.99 3654.98 3754.50 3802.73§
C 2629.45 2608.51 2550.45 2526.61 2577.17 2596.44§
µ 0.4 0.4 0.7 0.4 0.7 0.4§
____________________________________________________________________________
*Distances are in Å, angles in degrees, rotational constants in MHz,
dipole moments in Debye. **If no value is given in parentheses the
experimental error is less than the unit of the last digit given. §From
ref. [4]]. §§From ref. [16].
TABLE 7
Root mean square deviations of structural parameters and of rotational
constants calculated by various methods from their respective experimental
values (in percent).
_____________________________________________________________________
GAUSSIAN86 MOPAC
Molecule Parameter 6-31G* STO-3G MNDO MINDO/3 AM1
_____________________________________________________________________
Distances 1.02 1.14 0.80 1.70 1.08
Fulvene Angles 1.49 1.63 1.43 1.69 1.57
Rot. const. 1.22 0.62 1.92 2.73 0.88
Cyclopentadienone Rot. const. 1.44 0.90 2.16 1.28 2.05
4,4-DMCHDO* Rot. const. 0.81 1.42 2.13 2.66 0.70
1-MCHD** Rot. const. 0.90 0.98 1.44 2.39 0.97
Average§ Rot. const. 1.12 1.02 1.93 2.34 1.27
_____________________________________________________________________
*4,4-dimethyl-2,5-cyclohexadienone (IIc).
**1-methylene-2,5-cyclohexadiene (IIb). §Over the
rotational constants of all four molecules.
© Elsevier Science, 1994
All rights reserved. No part of this service may be reproduced, stored
in a retrieval system or transmitted in any form or by any means,
electronic, mechanical, photocopying, recording or otherwise, without
prior written permission of the publisher.
No responsibility is assumed
by Elsevier Science for any injury and/or
damage to persons or property as a matter of products liability,
negligence or otherwise, or from any use of operation of any methods,
products, instructions or ideas contained in the material herein.