Die verschiedenen Sätze von Parametern für die einzelnen Wechselwirkungen werden zu sogenannten Kraftfeldern zusammengefaßt [25]. Diese Kraftfelder sind für diverse Anwendungsgebiete, meist im makromolekularen Bereich, optimiert und je nachdem, welches Kraftfeld verwendet wird, erhält man zum Teil - im Maßstab der Genauigkeit der Mikrowellenspektroskopie - sehr unterschiedliche Strukturen.
Von diesen Programmpaketen standen zwei zur Verfügung, nämlich SYBYL [26] und DISCOVER/INSIGHT [27].
Im Laufe dieser Arbeit wurden drei verschiedene semi-empirische Methoden benutzt, nämlich MINDO/3 (modified intermediate neglect of differential overlap) [29], MNDO (modified neglect of diatomic overlap) [30] und AM1 (Austin model 1) [31]. Vor allem das AM1-Verfahren erwies sich in der Vorhersage von Strukturen und Dipolmomenten als recht brauchbar.
|
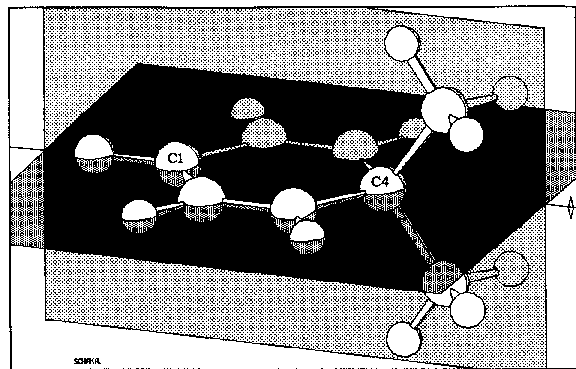
| Abb. C.1: Symmetrieelemente der betrachteten Verbindungen am Beispiel von DMCHDO. |
Alle diese Moleküle besitzen sehr ähnliche Symmetrie-Eigenschaften. Ein Symmetrieelement, das immer vorhanden ist, ist eine Spiegelebene senkrecht zum Ring, welche die Kohlenstoffatome C1 und, im Fall der Sechsringe, C4 enthält1 (vgl. Abb. C.1). Im Fall eines nicht-ebenen Ringes stellt diese Ebene das einzige Symmetrieelement dar und die resultierende Symmetriegruppe ist CS. Ist der Ring eben, dann bildet eben diese Ringebene eine zweite Symmetrieebene, die senkrecht auf der ersten steht. Dies bedingt gleichzeitig eine zweizählige Achse, die durch die Schnittlinie der beiden Spiegelebenen definiert wird. Die molekulare Symmetrie ist dann C2v.
Die Startparameter waren hier so gewählt, daß von einem gewinkelten Ring ausgegangen wurde. Demzufolge war als Symmetrie nur CS vorgegeben. Alle verwendeten Methoden ergaben als optimierte Struktur einen, im Rahmen der Abbruchkriterien, ebenen Ring, d.h. Diederwinkel innerhalb des Ringes von unter 0,1 Grad.
Es wäre zwar prinzipiell möglich gewesen, unter Vorgabe engerer Abbruchkriterien mit CS-Symmetrie eine genauere Struktur zu erhalten. Aufgrund der bis dahin erhaltenen Ergebnisse erschien es jedoch gerechtfertigt anzunehmen, daß das Potentialminimum einer Konfiguration mit C2v-Symmetrie entsprechen würde. So wurde eben diese Symmetrie im folgenden bereits fest vorgegeben, und die so erhaltene Struktur in einem neuen Programmlauf weiter optimiert. Auf diese Art und Weise konnte, vor allem bei den ab-initio-Rechnungen, viel Rechenzeit gespart werden. Die Ergebnisse sind in Tabelle C.1 zusammengefaßt.
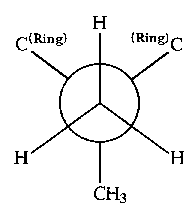 Da DMCHDO durch seine Größe schon einen erheblichen Rechenaufwand erfordert, wurde die interne Rotation der Methylgruppen "eingefroren". Dies erschien gerechtfertigt, da eine zuvor durchgeführte Analyse der Potentialhyperfläche bezüglich der Rotation der beiden Methylgruppen mit den zwei, oben angeführten, molecular-modelling-Programmen (SYBYL und DISCOVER/INSIGHT) Werte für die Rotationsbarriere von 17-20 kJ/mol vorausgesagt hatte. Es konnte also davon ausgegangen werden, daß sich die Methylgruppen im wesentlichen in den energetisch günstigsten Positionen befinden würden (vgl. Abb. C.2).
Da DMCHDO durch seine Größe schon einen erheblichen Rechenaufwand erfordert, wurde die interne Rotation der Methylgruppen "eingefroren". Dies erschien gerechtfertigt, da eine zuvor durchgeführte Analyse der Potentialhyperfläche bezüglich der Rotation der beiden Methylgruppen mit den zwei, oben angeführten, molecular-modelling-Programmen (SYBYL und DISCOVER/INSIGHT) Werte für die Rotationsbarriere von 17-20 kJ/mol vorausgesagt hatte. Es konnte also davon ausgegangen werden, daß sich die Methylgruppen im wesentlichen in den energetisch günstigsten Positionen befinden würden (vgl. Abb. C.2).
Obwohl die Ausgangskonformation eben war, wurde die Z-Matrix so aufgebaut, daß sie nur eine CS-Symmetrie (die "senkrechte" Spiegelebene) fixierte, während die Faltung des Ringes möglich war. Das Resultat der CS-Optimierung war bei allen Methoden eine ebene Ringstruktur und so wurde auch hier die Rechnung nochmals mit C2v-Symmetrie durchgeführt. Die Ergebnisse sind in Tabelle C.2 zusammengefaßt.
Das Molekül besitzt in dieser Konformation CS-Symmetrie. Diese wurde in der Z-Matrix fixiert und alle anderen Parameter zur Optimierung freigegeben. Bei Erreichen der Abbruchkriterien war die optimierte Struktur bei allen Methoden so flach (Diederwinkel unter 0,1 Grad), daß auch hier wieder die Struktur mit einer C2v-Symmetrie "verfeinert" wurde. Die Ergebnisse finden sich in Tabelle C.3.
Als Ausgangsstruktur wurde wieder die Wannen-Konformation verwendet, die auch als Startgeometrie für die Dimethyl-Methylen-Verbindung benutzt worden war. In der Z-Matrix wurden die beiden Methylgruppen hierzu einfach durch Wasserstoffatome ersetzt (C-H-Abstand 1,09 Å). Wiederum war die CS-optimierte Struktur bei allen Methoden effektiv eben. Es wurde deshalb wie bereits beschrieben mit C2v-Symmetrie optimiert. Die mit RHF/6-31G* so erhaltene Struktur ist mit der in [33] angegebenen identisch. Die Ergebnisse finden sich in Tabelle C.4.
Verläßliche Werte für Bindungsabstände und Winkel sind nur für Fulven verfügbar. Für dieses Molekül erhält man mit der MNDO-Methode die beste Übereinstimmung mit den experimentellen Strukturdaten. Jedoch sind die Abweichungen der anderen Methoden, insbesondere von RHF/6-31G* und AM1, nicht wesentlich
höher. Für die Rotationskonstanten von Fulven erhält man die besten Ergebnisse mit STO-3G. RHF/6-31G* und AM1 ergeben wiederum kaum schlechtere Resultate, während die mit MNDO berechneten Konstanten deutlich stärker von den experimentell ermittelten abweichen.
Ähnliche Ergebnisse erhält man auch für die Rotationskonstanten der anderen Moleküle, für die experimentelle Vergleichsdaten verfügbar sind: RHF/6-31G*, STO-3G (ab-initio) und AM1 (semi-empirisch) ergeben zwar nicht immer die kleinsten Abweichungen, sind aber im Mittel über alle Moleküle die zuverlässigsten Methoden. Zieht man weiterhin die benötigte Rechenzeit in Betracht, so erscheint AM1 die effektivste Methode zu sein, um Rotationskonstanten zu berechnen, während RHF/6-31G* bei der Berechnung von Molekülgeometrien eventuell die genaueren Daten liefert. Die Menge der Vergleichsdaten ist jedoch zu klein, um einen endgültigen Schluß zuzulassen.
_____________________________________________________________________________
GAUSSIAN86 MOPAC
6-31G* STO-3G MNDO MINDO/3 AM1
_____________________________________________________________________________
C1=O 1.199 1.227 1.228 1.207 1.239
C1-C2 1.482 1.511 1.498 1.508 1.474
C2=C3 1.324 1.317 1.349 1.349 1.339
C3-C4 1.499 1.520 1.506 1.498 1.483
C2-H 1.075 1.083 1.092 1.106 1.102
C3-H 1.077 1.085 1.093 1.108 1.102
C4-H 1.090 1.093 1.116 1.119 1.127
C2-C1=O 121.8 122.5 122.3 122.8 122.2
C2-C1-C6 116.4 114.9 115.5 114.4 115.6
C1-C2=C3 121.7 122.3 121.9 122.5 122.0
C2=C3-C4 123.3 123.7 123.6 123.5 123.3
C3-C4-C5 113.8 113.1 113.5 113.7 113.8
C1-C2-H 116.1 116.2 117.0 118.0 115.8
C3=C2-H 122.3 121.5 121.0 119.5 122.2
C2=C3-H 120.1 120.7 121.0 121.2 121.3
C4-C3-H 116.6 115.6 115.4 115.4 115.5
C3-C4-H 109.4 109.4 109.2 110.0 109.0
H-C4-H 105.1 106.0 106.4 102.8 106.8
A 5235.36 5128.09 5174.67 5198.95 5306.56
B 2721.84 2648.84 2615.96 2620.38 2650.41
C 1810.15 1765.36 1757.00 1760.95 1788.23
m 4.7 2.8 3.5 4.2 4.2
_____________________________________________________________________________
Tabelle C.1: Molekülstruktur von 2,5-Cyclohexadienon aus ab-initiound semi-empirischen Rechnungen. Hier, wie auch in den folgenden Tabellen, sind Abstände in Ångström, Winkel in Grad, Rotationskonstanten in MHz und Dipolmomente in Debye angegeben. Für dieses Molekül sind keine experimentellen Daten verfügbar.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.*
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=O 1.198 1.227 1.228 1.207 1.238
C1-C2 1.481 1.509 1.495 1.497 1.473
C2=C3 1.323 1.313 1.348 1.348 1.338
C3-C4 1.508 1.530 1.526 1.538 1.498
C2-H 1.075 1.083 1.092 1.106 1.102
C3-H 1.078 1.085 1.094 1.110 1.102
C4-C7 1.543 1.557 1.557 1.536 1.528
C7-H' 1.084 1.086 1.109 1.111 1.116
C7-H'' 1.085 1.086 1.109 1.111 1.116
C2-C1=O 122.0 122.7 122.5 123.3 122.3
C2-C1-C6 116.0 114.6 114.9 113.4 115.5
C1-C2=C3 121.6 122.4 122.3 123.0 122.2
C2=C3-C4 124.7 124.7 124.8 126.4 124.0
C3-C4-C5 111.5 111.2 110.8 107.9 112.3
C3=C2-H 122.2 121.5 120.7 118.8 122.0
C2=C3-H 119.7 120.3 119.7 119.3 121.3
C3-C4-C7 109.0 109.1 108.8 109.6 108.8
C4-C7-H' 110.8 110.6 111.2 113.5 110.4
C4-C7-H" 110.7 110.4 111.5 113.4 110.0
H'-C7-H" 108.1 108.4 107.6 105.2 108.8
H"-C7-H" 108.2 108.6 107.4 105.3 109.0
C7-C4-C8 109.3 109.1 110.8 110.4 109.2
A 3338.80 3276.33 3260.03 3306.67 3370.32 3332.16
B 1205.23 1178.95 1165.69 1151.86 1197.24 1193.07
C 1092.49 1067.37 1061.41 1050.04 1084.41 1082.21
m 4.8 2.9 3.6 4.3 4.2 4.5
____________________________________________________________________________
Tabelle C.2: Molekülstruktur von 4,4-Dimethyl-2,5-cyclohexadienon aus ab-initiound semi-empirischen Rechnungen im Vergleich mit den experimentellen Werten. Die Fehlergrenzen der experimentellen Werte sind kleiner als die Einheit der letzten angegebenen Ziffer. *Diese Arbeit.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.* [10]
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=C7 1.328 1.320 1.353 1.344 1.343 1.357**
C1-C2 1.472 1.491 1.477 1.493 1.459 1.478(6)
C2=C3 1.323 1.314 1.350 1.348 1.339 1.352(1)
C3-C4 1.512 1.530 1.525 1.539 1.498 1.493(6)
C2-H 1.077 1.083 1.093 1.108 1.102 1.075(5)
C3-H 1.078 1.084 1.093 1.109 1.101 1.075(5)
C4-C8 1.542 1.557 1.556 1.536 1.528 1.565(3)
C8-H' 1.084 1.086 1.109 1.112 1.116 1.175(4)
C8-H" 1.086 1.086 1.109 1.112 1.116 1.175(4)
C7-H 1.075 1.081 1.089 1.100 1.097 1.075(5)
C2-C1=C7 122.4 122.6 122.7 124.1 122.1
C2-C1-C6 115.3 114.8 114.6 111.8 115.7 115.7(3)
C1-C2=C3 122.4 122.7 123.1 124.3 122.4 120.8(3)
C2=C3-C4 124.6 124.5 124.5 126.1 123.7 125.6**
C3-C4-C5 110.7 110.8 110.3 107.5 112.0 110.5**
C3=C2-H 120.3 120.7 119.8 119.1 121.1 119.0**
C2=C3-H 119.5 120.3 119.8 119.0 121.4 119.5**
H'-C8-H" 108.1 108.4 107.5 105.1 108.8 110.0**
H"-C8-H" 108.1 108.5 107.3 105.0 108.9 110.0**
C8-C4-C9 109.1 109.0 110.7 110.1 109.0 112.5§
H-C7-H 116.7 116.2 113.2 110.1 115.6 121.0**§§
d$ 0.0 0.0 0.0 0.0 0.0 7.9(6)
A 3325.53 3266.14 3258.06 3300.81 3351.94 3229.59##
B 1195.20 1185.48 1161.36 1136.31 1200.43 1183.27##
C 1081.77 1071.21 1057.20 1035.90 1084.68 1082.05##
m 0.7 0.7 0.1 0.1 0.5
____________________________________________________________________________
Tabelle C.3: Molekülstruktur von 4,4-Dimethyl-1-methylen-2,5-cyclohexadien aus ab-initiound semi-empirischen Rechnungen im Vergleich mit den experimentellen Werten. *Abstände und Winkel sind als r(a)-Parameter angegeben. **Keine Fehler angegeben. §Angenommen. §§Aus dem Komplementwinkel berechnet. $Diederwinkel C6-C1-C2=C3. ##Aus der in [10] angegebenen Struktur berechnet.
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.*
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=C7 1.328 1.320 1.353 1.344 1.343
C1-C2 1.473 1.492 1.479 1.496 1.459
C2=C3 1.323 1.314 1.351 1.349 1.341
C3-C4 1.503 1.521 1.505 1.497 1.484
C2-H 1.076 1.083 1.093 1.108 1.102
C3-H 1.077 1.084 1.092 1.107 1.101
C4-H 1.090 1.093 1.116 1.120 1.127
C7-H 1.075 1.081 1.089 1.100 1.097
C2-C1=C7 122.2 122.4 122.4 123.6 122.0
C2-C1-C6 115.6 115.1 115.2 112.8 115.9
C1-C2=C3 122.4 122.6 122.7 123.8 122.2
C2=C3-C4 123.8 123.5 123.3 123.2 123.2
C3-C4-C5 112.8 112.5 112.9 113.2 113.5
C1-C2-H 117.3 116.6 117.2 116.4 116.5
C3=C2-H 120.3 120.7 120.2 119.8 121.3
C2=C3-H 119.8 120.6 121.0 120.7 121.3
C4-C3-H 116.8 115.8 115.6 116.0 115.6
C3-C4-H 109.7 109.6 109.4 110.2 109.1
H-C4-H 105.1 105.7 106.2 102.4 106.8
C1-C7-H 121.7 121.9 123.4 125.0 122.2
H-C7-H 116.7 116.2 113.1 110.0 115.6
A 5194.13 5100.58 5167.70 5181.15 5254.47 5177.82
B 2645.62 2634.14 2559.81 2523.74 2621.12 2613.15
C 1771.37 1755.54 1730.67 1714.78 1768.95 1755.84
m 0.8 0.6 0.1 0.1 0.5 0.86
____________________________________________________________________________
Tabelle C.4: Molekülstruktur von 1-Methylen-2,5-cyclohexadien aus ab-initiound semi-empirischen Rechnungen im Vergleich mit den experimentellen Werten. Die Fehlergrenzen der experimentellen Werte sind kleiner als die Einheit der letzten angegebenen Ziffer. *Diese Arbeit.
______________________________________________________________________________
GAUSSIAN86 MOPAC exp.*
6-31G* STO-3G MNDO MINDO/3 AM1
______________________________________________________________________________
C1=C6 1.325 1.319 1.345 1.338 1.332 1.3485(5)§
C1-C2 1.477 1.495 1.491 1.507 1.483 1.468(1)$
C2=C3 1.333 1.324 1.366 1.359 1.363 1.357(1)$
C3-C4 1.480 1.492 1.476 1.480 1.476 1.476(2)§
C2-H 1.073 1.080 1.082 1.100 1.088 1.078(1)§
C3-H 1.073 1.081 1.082 1.100 1.088 1.080(1)§
C6-H 1.076 1.083 1.089 1.101 1.098 1.077(1)$
C2-C1=C6 127.3 127.8 127.8 128.5 127.4
C2-C1-C5 105.5 104.4 104.4 103.0 105.2 106.6(1)§
C1-C2=C3 108.2 108.6 109.0 109.7 108.6 107.7(1)§
C2=C3-C4 109.1 109.2 108.8 108.9 108.8 109.0(2)§
C1-C2_H 124.3 123.5 123.4 122.7 122.9 124.7(2)§
C3=C2-H 127.5 127.9 127.7 127.7 128.6
C2=C3-H 126.6 127.2 127.7 127.7 128.0 126.4(2)§
C4-C3-H 124.3 123.6 123.5 123.4 123.1
C1-C6-H 121.7 122.0 123.2 125.1 122.2 126.4(1)$
H-C6-H 116.6 116.0 113.6 109.9 115.7
A 8240.05 8152.65 8223.85 8184.11 8218.53 8186.13§
B 3861.77 3835.81 3696.99 3654.98 3754.50 3802.73§
C 2629.45 2608.51 2550.45 2526.61 2577.17 2596.44§
m 0.4 0.4 0.7 0.4 0.7 0.4§
______________________________________________________________________________
Tabelle C.5: Molekülstruktur von Fulven aus ab-initiound
semi-empirischen Rechnungen im Vergleich mit den experimentellen Werten.
Falls kein Fehler angegeben ist, sind die Fehlergrenzen der experimentellen
Werte kleiner als die Einheit der letzten angegebenen Ziffer. *Abstände
und Winkel sind als rs-Struktur angegeben. §Aus [34a]. $Aus [34b].
____________________________________________________________________________
GAUSSIAN86 MOPAC exp.
6-31G* STO-3G MNDO MINDO/3 AM1
____________________________________________________________________________
C1=O 1.188 1.220 1.218 1.198 1.224
C1-C2 1.506 1.525 1.518 1.521 1.509
C2=C3 1.322 1.317 1.358 1.352 1.353
C3-C4 1.503 1.506 1.489 1.494 1.490
C2-H 1.071 1.080 1.081 1.098 1.088
C3-H 1.073 1.082 1.083 1.101 1.090
C2-C1=O 127.3 128.0 127.8 127.7 127.7
C2-C1-C5 105.5 104.0 104.4 104.6 104.7
C1-C2=C3 107.5 108.1 108.2 108.0 108.3
C2=C3-C4 109.8 109.9 109.6 109.7 109.4
C1-C2-H 123.5 123.5 123.1 124.6 122.1
C3=C2-H 129.0 128.5 128.6 127.4 129.6
C2=C3-H 127.0 127.4 127.9 128.1 128.4
C4-C3-H 123.2 122.7 122.6 122.1 122.2
A 8216.82 8170.95 8239.60 8161.31 8287.97 8152.17*
B 4024.21 3902.38 3831.37 3880.45 3843.75 3939.56*
C 2701.26 2641.04 2615.28 2629.97 2625.92 2655.80*
m 3.6 1.9 2.8 3.4 2.9 3.1§
____________________________________________________________________________
Tabelle C.6: Molekülstruktur von Cyclopentadienon aus ab-initiound semi-empirischen Rechnungen im Vergleich mit den experimentellen Werten. Die Fehlergrenzen der experimentellen Werte sind kleiner als die Einheit der letzten angegebenen Ziffer. *Aus [35a]. §Aus [35b].
_______________________________________________________________________
GAUSSIAN86 MOPAC
Molekül Parameter 6-31G* STO-3G MNDO MINDO/3 AM1
_______________________________________________________________________
/Abstände 1.02 1.14 0.80 1.70 1.08
Fulven -( Winkel 1.49 1.63 1.43 1.69 1.57
\Rot. konst. 1.22 0.62 1.92 2.73 0.88
Cyclopentadienon Rot. konst. 1.44 0.90 2.16 1.28 2.05
4,4-DMCHDO Rot. konst. 0.81 1.42 2.13 2.66 0.70
1-MCHD Rot. konst. 0.90 0.98 1.44 2.39 0.97
Durchschnitt* Rot. konst. 1.12 1.02 1.93 2.34 1.27
_______________________________________________________________________
Tabelle C.7: Durchschnittliche Abweichung der Strukturparameter und der Rotationskonstanten, die mit  Zurück zum Inhaltsverzeichnis
Zurück zum Inhaltsverzeichnis